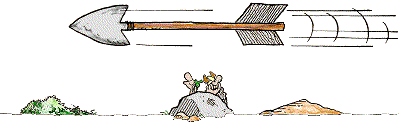
red square: 1 mm2 = 100 nl
green square: 0.0625 mm2 = 6.25 nl
yellow square: 0.04 mm2 = 4 nl
blue square: 0.0025 mm2 = 0.25 nl
depth: 0.1 mm
CELL LINES USED IN THE CBL
Based on hteir morphology and/or functions 3 types of cell lines can be differenciated:| Cell line | Description | Medium | Type | Inoculum | Subculture | Freeze | BSL | ATCC |
| HeLa | Human cervix (epithelial) | DMEM/F12 + 10% FBS | adherent | 1 * 104/cm2 | 1:10 | 75% DMEM/F12, 20% FBS, 5% DMSO | 2 (HPV-18) |
CCL-2 |
| Caco-2 | Human colon (epithelial-like) | DMEM/F12 + 10% FBS | adherent | 1 * 104/cm2 | 1:10 | 75% DMEM/F12, 20% FBS, 5% DMSO | 1 |
HTB-37 |
| MCF7 | Human breast (epithelial) | EMEM + 10% FBS + 0.01 mg/ml insulin or DMEM/F12 + 10% FBS + 0.01 mg/ml insulin ? |
adherent @ 37 °C not adherent @ RT |
1 * 104/cm2 | 1:10 | 85% EMEM, 10% FBS, 5% DMSO | 1 | HTB-22 |
| HT-29 | Human colon (epithelial-like) | DMEM + 10% FBS | adherent | 5 * 103/cm2 | 1:20 | 75% DMEM, 20% FBS, 5% DMSO | 1 |
HTB-38 |
| THLE-3 | Human liver (epithelial) | WME-L + 10% FBS | adherent | 104/cm2 coated flask |
1:10 | 75% WME-L, 20% FBS, 5% DMSO | 2 (SV40) |
CRL 11233 |
| THP-1 | Human monocytes (monocyte) | RPMI1640 or DMEM/F12 + 10% FBS | suspension | --- | 1:10 | 75% medium, 20% FBS, 5% DMSO | 1 |
TIB 202 |
|
Improved Neubauer-Kammer red square: 1 mm2 = 100 nl green square: 0.0625 mm2 = 6.25 nl yellow square: 0.04 mm2 = 4 nl blue square: 0.0025 mm2 = 0.25 nl depth: 0.1 mm |
|
| T-75 | T-25 | 6-well MTP | 12-well MTP | 96-well MTP | |
| working vol. (ml) | 15 | 5 | 2.5 | 1 | 0.1 |
| working surface (cm2) | 75 | 25 | 9.5 | 4 | 0.32 |
| Total cells (N) at 100% confluence | 8 * 106 | 3 * 106 | 1 * 106 | 4 * 105 | 3.5 * 104 |
| Wash DPBS (ml) | 8 | 3 | 1.5 | 0.5 | 0.1 |
| Trypsin/EDTA (ml) | 1.5 | 0.5 | -- | -- | -- |
| Add new medium (ml) | 7.5 | 2.5 | -- | -- | -- |
| Expected cells (N/ml) | 1 * 106 | 1 * 106 | -- | -- | -- |
| Needed cells (N): 4d subculturing in 2 days 70% confluent next day 70% confluent |
5.0 * 105 -- -- |
2.0 * 105 -- -- |
-- 1.75 * 105 3.5 * 105 |
-- 7.0 * 104 1.4 * 105 |
-- 6.0 * 103 1.2 * 104 |
| Expected inoculum: 4d subculturing in 2 days 70% confluent next day 70% confluent |
500 µl -- -- |
200 µl -- -- |
-- 175 µl 350 µl |
-- 70 µl 140 µl |
-- 6 µl 12 µl |
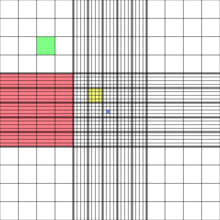
| slow cooling | fast cooling |
| external water freezes first | internal water freezes first |
| -> water migrates out of the cells (osmosis) | -> water remains in the cells |
| -> avoids formation of intracelular ice crystals | -> avoids osmotic imbalance |
| => but high osmotic imbalance | => but formation of ice crystals |
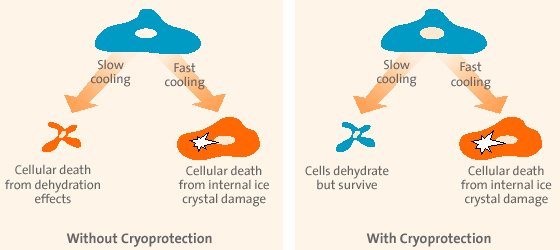
| Seed to ~40% confluent 1 d before transfection i.e. 2.5 * 105 N for 30 mm culture dish (= 250 µl) or 8 * 105 N for T-25 flask (= 800 µl) Incubate cells 24 h Mix 2 / 5 µg DNA (~1 µg/µl) with 200 / 500 µl serum-free DMEM (vortex). - add 2 / 5 µl (vortexed) PolyMAG, vortex 10 s strongly. Incubate 20 min at RT. - add mixture to the cells (30 mm culture dish or T-25 flask) - gently rock for even distribution - incubate 20 min on Magnetofector-plate in the incubator (not longer!) Incubate cells 24-48 h. |
|
| Basic vector | Derivates | Promotor | Features | Resistance gene | Antibiotic | Conc: selection / maintenance |
| pcDNA3.1 (Invitrogen) |
pDHH-... p...-HH pDGM-... p...-GFP |
pCMV | His/Xpress-MCS-GENE GENE-MCS-His/V5 GFP-MCS-GENE GENE-MCS-GFP |
NEOR (APHR, KANR) | G418 = Genticin Stock: 50 mg/ml (HEPES) store: -20 °C |
500 µg/ml / 100 µg/ml add: 10 µl/ml / 2 µl/ml |
| pcDNA3.1/Hygro (Invitrogen) |
p..._Hyg | pCMV | MCS-GENE | HPHR (HYGR) | Hygromycin B Stock: 40 mg/ml (HEPES) store: 4 °C |
200 µg/ml / 100 µg/ml add: 5 µl/ml / 2.5 µl/ml |
| pTet-On1 (Clonetech) |
pPEx, pHAPEx pPSelGFP |
pCMV | GENE-MCS | NEOR (APHR, KANR) | G418 = Genticin Stock: 50 mg/ml (HEPES) store: -20 °C |
500 µg/ml / 100 µg/ml add: 10 µl/ml / 2 µl/ml |