SEPARATION OF PROTEINS UNDER DENATURATING CONDITIONS IN AN ELECTRIC FIELD.
The process can be divided into 4 steps:| Use Nitril gloves to: - not transfer fat or proteins into the experiment - protect yourself against acrylamid (> 480 min) beware: they do not protect againt acetone! |
 |
| 1) Clean glass plates with acetone (fatt-free) and let them dry 2) Place Short Plate on top of Spacer Plate (1 mm) Top side of Spacer Plate is indicated 3) Slide both into the green Casting Frame Keep the Short Plate in front Insure that both plate reach the bottom and align them perfectly 4) Lock the pressure clamps to secure glass plates Place a strip of parafilm on the bottom of the glass plates to avoid leaking of unpolymerized acrylamide |
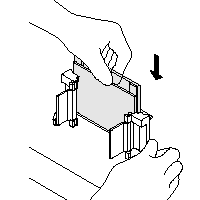 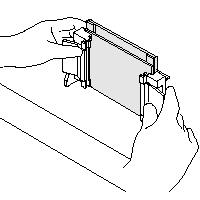 |
| 5) Place grey Gasket (rubber-foam) onto the Casting Stand Keep Gasket clean and dry, as it swells when soaked with water (if so, let them air dry before gel casting) 6) Place Casting Frame on Casting Stand with engaged clamps in front Fix Plates/Casting Frame with the spring lever 7) Insure the save standing and avoid any misaligning (images are taken from the instruction manual) |
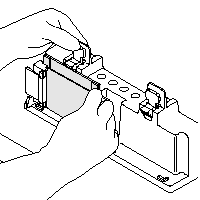 |
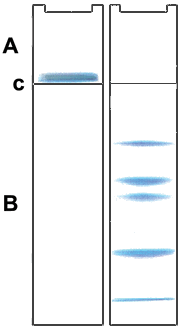
| The Discontinuous or Laemmli or Tris-Glycine-Buffer System separates the denaturalized protein-SDS complex by molecular sieve effect in a polyacrylamid matrix. The pore size of the matrix determines the separation efficiency and depends on the molecular weight of the proteins. Usually the Laemmli gels are composed of an upper stacking or casting gel (A), to concentrate the protein sample, and a resolving or separating gel (B), to separate the proteins by their molecular weight. Like this the sample is concentrated at the border between both (C), the proteins migrate more homogeneously into the resolving gel and thus sharper band can be achieved. (image taken from "wikipedia") |
 |
| Gel | Stacking (3 ml) | Resolving (7 ml) | |||
| Conc. | 5 % | 6 % | 8 % | 10 % | 12 % |
| A. dest. [ml] | 2.185 | 4.06 | 3.71 | 3.36 | 3.01 |
| 40% Acrylamid/bis, 37,5:1 [ml] | 0.375 | 1.05 | 1.40 | 1.75 | 2.10 |
| Resolving Gel Buffer 1.5 M Tris, pH 8.8 [ml] |
-- | 1.75 | 1.75 | 1.75 | 1.75 |
| Stacking Gel Buffer 0.5 M Tris, pH 8.8 [ml] |
0.38 | -- | -- | -- | -- |
| 10% SDS [µl] | 30 | 70 | 70 | 70 | 70 |
| degas the mixture for 15 min | + | + | + | + | + |
| TEMED [µl] add prior pouring gel (Biorad recommends 5 µl for resolving gel?) |
3 | 7 | 7 | 7 | 7 |
| 10 % (fresh!) APS [µl] add prior pouring gel |
30 | 70 | 70 | 70 | 70 |
| 1) Remove Casting Frame from Casting Stand 2) Release PA-gel Cassette from Casting Frame NOTE: When running 1 or 2 gels, use the Assembly Module (the one with the banana plugs), only when running 3 or 4 gels, use additionally the Companion Module. 3) Open the lateral green clamps from the Module and place it on the desk 4) Place one PA-gel Cassette onto the white support, the short plate facing inwards Place on the counter-side a second PA-gel Cassette (short plate facing inwards) or the dummy-plate (buffer-dam) |
  |
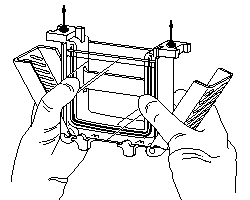
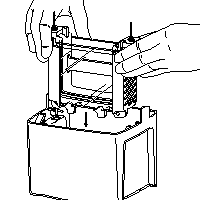
| 5) Keep both Cassettes upright with the hands and align them perfectly Make sure that the short plate sits below the notch of the green seal Close the lateral green clamps to fix the plates 6) Place the Module(s) into the Tank Beware the orientation: black (-) and red (+) NOTE: When running only 1 or 2 gels DO NOT place the (empty) Companion Module into the Tank! 7) Add ~150 ml Running Buffer between the plates (Upper Chamber) just under the top of the Spacer Plate (outer gel plate) 8) Remove carefully the comb and wash the wells with Running Buffer in order to remove residual, not polymerized acrylamide 9) Remove all formed air bubble (in the wells or under the gel) (images are taken from the instruction manual) |
  |
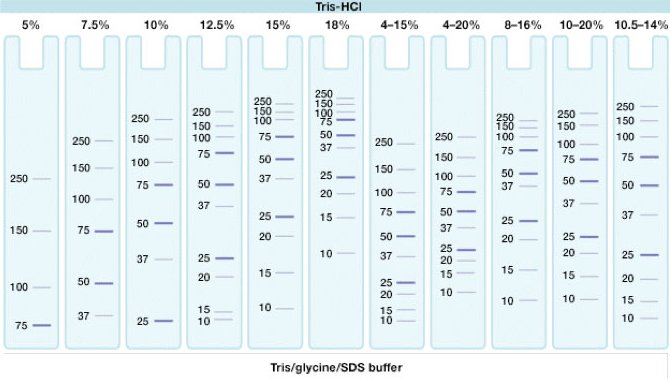
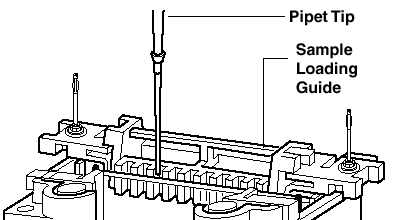
| 1) Place the (yellow) Loading Guide between the 2 gels press/fix the Loading Guide against one plate at the time? 2) Use the slot to load the samples into the corresponding wells (pipette) Load samples slowly to led them settle down Use same volumina to avoid distortions For the same reason fill not used wells with 1X Sample/Loading Buffer Load 5-10 µl Rainbow Size Marker (Biorad #161-0324) into one well of each gel Protein load: well with 1 mm (thick) x 5 mm (wide): up to 40 µl sample and 0.6 mg protein 3) Fill the tank with Running Buffer until the indicated level: 550 ml for 1-2 gels and 680 ml for 3-4 gels 4) Tap the tank and insert the cable into a Power supply (Major Science, MP-3AP) 5) Use constant 200 V (for 1-2 as well as for 3-4 gels), 60-80 mA at begin, 40 mA at the end Run time is about 60 min use 10 mA until bromphenol blue reached resolving gel, then use 15 mA Run time is about 60-90 min use 140 V; at the beginning 100-125 mA, at the end 60-80 mA Run time is about 55 min |
 |
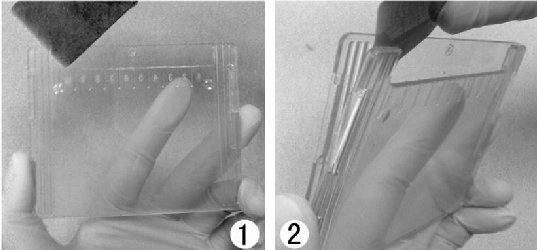
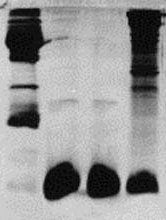
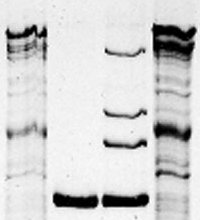
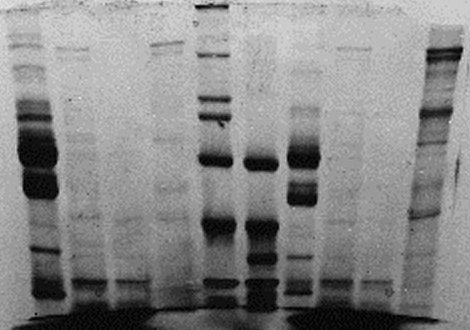
| 6) Turn off power when bromphenol blue has reached the end of the resolving gel and disconnect tank 7) Remove lid, take out Module(s) Pour off Running Buffer from the Module (Upper Chamber) 8) Open lateral green clamps from the Module and remove the PA-gel Cassette Separate the glass plates by introducing a green Gel Releaser and twist carefully Float the gel off, e.g. with A. dest. |
 |
 |
 |
 |
 |
 |