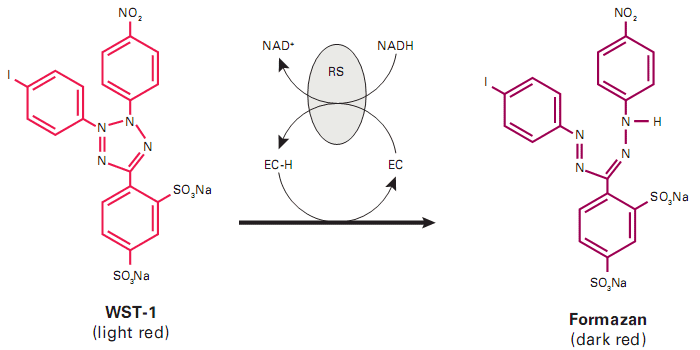
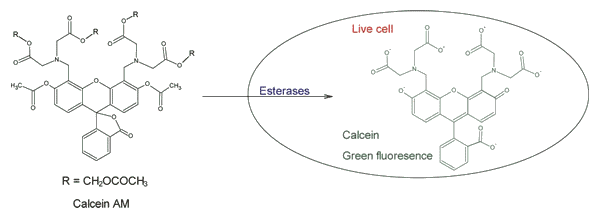
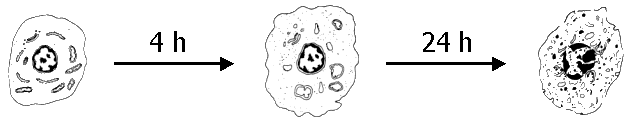
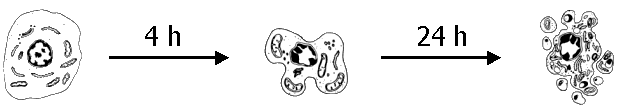
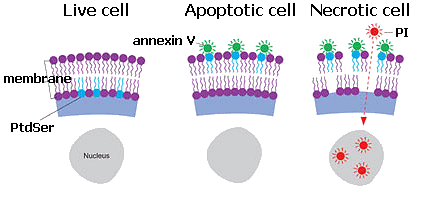
- use of human cells (and not from animals)
- reduce the number of animal 'consuming' experiments
- cellular effects can be detected
- automatization is possible (high throughput screening)
as well the disadvantages are:
- systemic effects are not (or only difficult) detectable
- no long-term studies
- metabolization of components can only be simulated
- a correlation between the in-vitro and in-vivo results is required
Finally, experiments with animals can not be substituted but at least reduced.

This is still not possible.